Research

Transport and Dispersion of Exciton-Polaritons
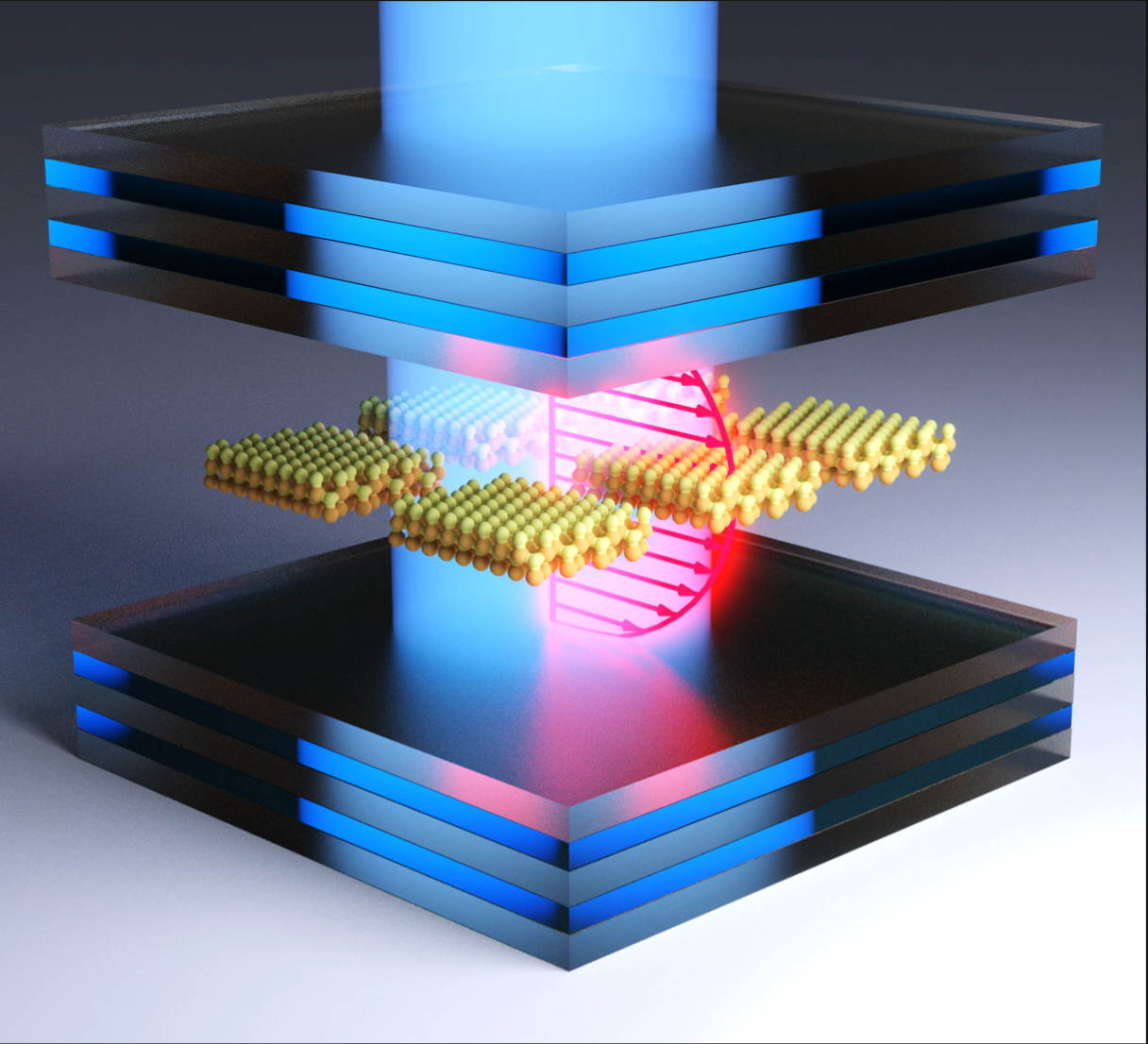
Outside the cavity, materials exhibit diffusive excitonic transport due to strong exciton-phonon interactions, which induce dynamic disorder. We found that when placing materials inside an optical cavity, light-matter interactions can strongly modify transport properties by effectively modifying exciton-phonon interactions. We observe that exciting to a polaritonic branch with high photonic character leads to ballistic propagation with velocities that match the expected group velocity from polaritonic dispersion. This is because polariton branches with high photonic character couple weakly to the phonons, thereby having a long decoherence time. When initial excitation is of higher excitonic character, we observe ballistic motion with velocities that are smaller than expected group velocities from polaritonic dispersion. With our direct quantum dynamics simulations, we show that this is due to a transient localization mechanism. We also developed a microscopic theory to obtain multimode polariton dispersion in materials coupled to cavity radiation modes. Using this theory, we shed light on the ambiguity of using a 2N or N+1 model for obtaining dispersion and developed a general strategy for obtaining simple matrix models based on the structure and spatial location of multi-layered 2D materials inside the optical cavity.
- Xu† Mandal† Baxter Cheng Lee Su Liu Reichman* Delor* Nature Comm. (2023)
- Mandal* Xu Mahajan Lee Delor Reichman* Nano Lett. (2023)

New quantum dynamics approach for on-the-fly excited state dynamics: Quasi-Diabatic propagation scheme
Direct on-the-fly quantum dynamics simulation requires combining quantum dynamics methods with electronic structure approaches. However, they are usually formulated under two different representations. While many quantum dynamics methods are developed in the diabatic representation, most electronic structure approaches provide outputs in the adiabatic representation. To address this discrepancy, we have worked to develop the quasi-diabatic (QD) propagation scheme, which uses the adiabatic states as the quasi-diabatic states (or local diabatic states) during a short-time propagation and continuously updates the QD basis at each consecutive nuclear propagation step. The QD scheme allows the use of any diabatic dynamics approach to propagate on-the-fly quantum dynamics using any adiabatic electronic structure method.
- Mandal† Yamijala† Huo* J. Chem. Theory Comput. (2018)
- Mandal Shakib* Huo* J. Phys. Chem. (2019)
- Zhou Mandal Huo* J. Phys. Chem. Lett. (2019)
Investigating New chemical reactivities in Polariton Chemistry
In polaritonic chemistry, the quantum nature of photons is harnessed to enable new chemical reactivities. Through direct quantum-dynamics simulations and analytical theories, we investigate new chemical reactivities enabled by coupling quantized radiation to molecular systems. With these tools, we provide time-dependent mechanistic insights and discover new ways of enabling and controlling chemical reactivities. We have investigated the possibility of tuning an isomerization reaction, suppressing or enhancing electron transfer rates and demonstrated that molecule-cavity setups can be used to do down-conversion.
- Mandal Huo* J. Phys. Chem. Lett. (2018)
- Mandal* Krauss* Huo* J. Phys. Chem. B (2020)

New Theoretical Framework for Molecular Quantum Electrodynamics
We have developed a new theoretical framework that resolves the gauge ambiguity, i.e. under electronic state truncation (few-level description of matter), the dipole gauge Hamiltonian provides a completely different result than the Coulomb gauge Hamiltonian. While the dipole gauge Hamiltonian (so called d.E) provides accurate results , the Coulomb gauge Hamiltonian (so called p.A) breaks down, especially in the ultra-strong coupling regime. We derived two new Coulomb gauge Hamiltonians that provide consistent results compared to the d.E Hamiltonian. At the same time, these new Hamiltonians provide computational convenience as they require relatively fewer Fock states compared to the dipole-gauge Hamiltonian and relatively fewer electronic states compared to the Coulomb gauge Hamiltonian for converging results. Using the same approach, we have also resolved ambiguities arising when performing mode truncation, i.e. describing light-matter hybrid systems with only one/few cavity modes.
- Taylor† Mandal† Zhou Huo* Phys. Rev. Lett. (2020)
- Mandal* Vega Huo* J. Phys. Chem. Lett. (2020)
- Taylor Mandal Huo* Opt. Lett. (2022)

Chemical Kinetics under Vibrational Strong Coupling
Recent experiments have demonstrated that ground state chemical kinetics can be modified by coupling molecular vibrations to quantized radiation in an optical cavity. The theoretical understanding of this remarkable effect remains elusive. We have developed a series of theories, that demonstrate that the radiation modes can act as non-Markovian 'solvent' degrees of freedom, effectively enhancing environmental friction experienced by molecules. In the classical treatment, we showed that the cavity can either enhance or suppress chemical reactivity depending on whether the solvent coupling (or equivalently friction) places the system in the energy diffusion-limited (weak-coupling) or spatial diffusion-limited regime. However, our classical theories predict a much broader range of cavity frequency dependence of chemical reactivity than is observed in experiments. Finally, with exact quantum dynamics simulations, we discovered that cavities can resonantly either suppress or enhance chemical reactivity depending on a multitude of factors, such as cavity loss, cavity coupling strength, solvent friction, shape of the solvent spectral density, details of the vibrational eigenstates of the molecule, etc. The quantum results show much more visually similar (sharp) cavity frequency dependence of chemical kinetics modulation under vibrational strong coupling compared to the experiments.
- Lindoy Mandal Reichman* Nature Comm. (2023)
- Li Mandal* Huo* Nature Comm. (2021)
- Mandal* Li Huo* J. Chem. Phys. (2022)
- Li* Mandal* Huo* J. Phys. Chem. Lett. (2021)
- Li Mandal Reichman* J. Phys. Chem. Lett. (2022)